

Patologie chromosomowe są jednym z najpoważniejszych zaburzeń w komórkach, z powodu podwojenia jednej z par chromosomów, pojawiają się specjalne zespoły, które mają typowe zewnętrzne objawy i zmiany w metabolizmie. Trisomia w jednej z par chromosomów (gdy zamiast dwóch są trzy chromosomy) występuje najczęściej w trzech wariantach - zespole Downa, który jest najczęstszy i powszechnie znany, a także Patau i Edwards. Otrzymali swoje nazwiska ze względu na ich odkrycie i opis przez naukowców..
Dane ogólne
Zespół Edwardsa (lub zespół trisomii 18) jest na drugim miejscu pod względem występowania trisomii, po zespole Downa, i występuje, gdy 18. para chromosomów nie pęka. Patologia charakteryzuje się różnymi zaburzeniami w tworzeniu się układów płodowych i narządów w macicy, ma typowe zewnętrzne zmiany i niekorzystne prognozy zdrowotne i życiowe.
Jest ważnyDzieci należą do kategorii osób ciężko niepełnosprawnych, wymagają szczególnego traktowania, opieki i opieki nad swoimi rodzicami i lekarzami..
 Pod względem częstości występowania ta nieprawidłowość chromosomowa jest uważana za dość częstą, do 0,01-0,02% dzieci urodzonych na świecie ma tę nieprawidłowość chromosomową., i brak zależności od konkretnej rasy lub kraju zamieszkania, jest wszechobecny. Według statystyk dziewczęta cierpią 3-4 razy częściej niż chłopcy, chociaż nie ma dobrze uzasadnionych danych naukowych dotyczących tego zjawiska. Dokładna przyczyna powstawania trisomii u płodu nie została ustalona, ale zidentyfikowano wiele specyficznych czynników ryzyka, które zwiększają prawdopodobieństwo jej rozwoju..
Pod względem częstości występowania ta nieprawidłowość chromosomowa jest uważana za dość częstą, do 0,01-0,02% dzieci urodzonych na świecie ma tę nieprawidłowość chromosomową., i brak zależności od konkretnej rasy lub kraju zamieszkania, jest wszechobecny. Według statystyk dziewczęta cierpią 3-4 razy częściej niż chłopcy, chociaż nie ma dobrze uzasadnionych danych naukowych dotyczących tego zjawiska. Dokładna przyczyna powstawania trisomii u płodu nie została ustalona, ale zidentyfikowano wiele specyficznych czynników ryzyka, które zwiększają prawdopodobieństwo jej rozwoju..
Wraz z innymi zaburzeniami chromosomalnymi i genowymi, dzisiaj zespół Edwardsa obejmuje wrodzone i nieuleczalne patologie, specjalne metody opieki i wsparcie medyczne, środki rehabilitacyjne pozwalają w określony sposób wspierać życie dzieci i budować sukces w ich rozwoju - fizycznym i psychicznym. Ale ze względu na ogromną różnorodność zaburzeń, jakie daje dodatkowy chromosom, nie ma jednolitych metod leczenia i opieki.
Historia badań patologii
W medycznych traktatach i fikcji można znaleźć pojedyncze informacje na temat pacjentów z tym zespołem w czasach starożytnych, ale zespół został szczegółowo opisany na początku ubiegłego wieku. Rozwój technologii czasu i wcześniej nie pozwalał na szczegółowe zbadanie tej patologii, więc przyczyny tego nie wiedziały. Ponadto większość dzieci urodzonych z tym zespołem zmarło w młodym wieku z powodu słabego rozwoju medycyny i udzielania im na czas pomocy. Przyczyna zespołu - trisomia na 18. parze chromosomów została odkryta dopiero w 1960 r. Przez D. Edwardsa, a nowa patologia została nazwana jego imieniem. Zgodnie ze statystykami medycznymi występowanie patologii wynosi średnio 1 przypadek na 3000 koncepcji, ale przed urodzeniem jest znacznie mniej płodów. To dlatego, że podobna anomalia często na samym początku kończy się poronieniem lub śmiercią płodów w macicy. Ale często, przy takich poronieniach, nie są wykonywane żadne badania chromosomalne..
Czym jest trisomia
Mówiąc z punktu widzenia medycyny, trisomia jest odmianą mutacji chromosomowych, gdy komórki pacjenta nie mają 46 chromosomów złożonych w homologiczne 23 pary, z których jedna określa różnice płciowe, ale wykryto 47 chromosomów.
Dzisiaj lekarze rozróżniają trzy rodzaje trisomii, które należą do nieśmiercionośnych mutacji - zespół Downa (cierpi 21 para), zespół Patau (cierpi 13. para) i zespół Edwardsa (uszkodzenie wykryto u 18-mej pary). Wszystkie inne warianty trisomii są początkowo niezgodne z życiem, takie zarodki natychmiast umierają, a ciąża nie rozwija się. Ale narodziny dzieci z tymi trzema trisomią są zawsze problemem ich zdrowia, rozwoju fizycznego i inteligencji, funkcji umysłowych..
Przyczyny zespołu Edwardsa
Podobnie jak w przypadku pozostałych dwóch wyżej wymienionych patologii, zespół Edwardsa określa się jako patologie genetyczne, w których w autosomach znajduje się dodatkowy chromosom (nie chromosomów płciowych). Osoba zdrowa ma 46 chromosomów - 22 pary - autosomów (nie-seksu) i jedną parę - płeć, określającą, czy jest to dziewczyna czy chłopak. Nici DNA są zwarte w chromosomach, w których zapisywane są wszystkie informacje genetyczne dotyczące organizmu, począwszy od procesu poczęcia aż do śmierci. Cały program życia zapisany jest w niciach DNA..
W zespole Edwardsa organizm nie cierpi na defekty genu, ale na chromosomalny występuje defekt w podziale komórki zarodkowej, który następnie daje dziecku życie, a dodatkowy chromosom pozostaje w nim. W klasycznej wersji powstawania zespołu Edwardsa występuje podwojenie 18 chromosomu i zarówno chłopiec, jak i dziewczynka mogą urodzić się z podobnym zespołem.. Nadmiar informacji genetycznej znajduje się we wszystkich komórkach takiego dziecka. Taka nadmiarowość DNA prowadzi do szeregu zaburzeń, które prowadzą zarówno do zewnętrznych defektów, jak i problemów w pracy układów i narządów. Ze względu na obecność trzeciego, dodatkowego chromosomu, pojawiła się nazwa naukowa - trisomia.
Rodzaje zespołu Edwardsa: jak zależy od nasilenia objawów
W zależności od rodzaju defektu chromosomowego występującego w zespole istnieją trzy rodzaje przepływu:
- Całkowita trisomia 18 pary.
- Częściowa trisomia 18 pary
- Mozaika w formie patologii.
Dzięki pełna forma (należy również do klasycznego syndromu) zakłada się, że całkowicie we wszystkich komórkach występuje dodatkowy chromosom. Ta opcja jest najtrudniejsza i najczęściej występuje, prognozy z nią są najbardziej niekorzystne..
 Częściowa trisomia odnosi się do rzadkich wariantów patologii, występuje w 3% przypadków, przy czym ten problem nie zawiera pełnego trzeciego chromosomu, a jedynie jego fragment. Taki defekt może wystąpić w wyniku problemów z podziałem komórek (zwłaszcza ich chromosomów), co jest niezwykle rzadkie. Może to być również wariant tego, że dodatkowa część 18-go chromosomu jest przyłączona do poszczególnych cząsteczek DNA, co prowadzi do ich wydłużania i zmiany. W tych komórkach znajdują się dwa 18-sze chromosomy i część nadmiaru materiału. Wtedy zespół wrodzony nie będzie tak ciężki, ponieważ dodatkowe informacje pojawiają się w komórkach nie wszystkich informacji, ale tylko ich części.. Prognozy dla tego stanu, choć niekorzystne, są dużo łatwiejsze dla zespołu niż dla pełnej postaci..
Częściowa trisomia odnosi się do rzadkich wariantów patologii, występuje w 3% przypadków, przy czym ten problem nie zawiera pełnego trzeciego chromosomu, a jedynie jego fragment. Taki defekt może wystąpić w wyniku problemów z podziałem komórek (zwłaszcza ich chromosomów), co jest niezwykle rzadkie. Może to być również wariant tego, że dodatkowa część 18-go chromosomu jest przyłączona do poszczególnych cząsteczek DNA, co prowadzi do ich wydłużania i zmiany. W tych komórkach znajdują się dwa 18-sze chromosomy i część nadmiaru materiału. Wtedy zespół wrodzony nie będzie tak ciężki, ponieważ dodatkowe informacje pojawiają się w komórkach nie wszystkich informacji, ale tylko ich części.. Prognozy dla tego stanu, choć niekorzystne, są dużo łatwiejsze dla zespołu niż dla pełnej postaci..
Rozwój kształt mozaiki pojawia się u około 5-8% dzieci z podobną anomalią. Ale mechanizmy tego rodzaju syndromu są różne. Co ciekawe, podobna forma rozwija się po połączeniu się plemnika z komórką jajową, a obie uzyskane komórki początkowo mają normalny zestaw chromosomów - 46XX lub 46XY. Ale na pierwszych podziałach wykształconej zygoty powstała usterka, a jedna z komórek nabyta dzięki temu dodatkowemu chromosomowi. W rezultacie zarodek nabył komórki z prawidłowymi chromosomami - jest ich 46, druga część ma dodatkowy chromosom - 47. Liczba komórek z dodatkowym chromosomem z takimi problemami nie przekracza 50%.
Zwróć uwagęIm wyższa liczba wadliwych komórek w zarodku i dziecku, tym gorsze prognozy i ich objętość zależy od czasu, w którym doszło do awarii podczas dzielenia. Im później to nastąpiło, tym mniejsza objętość wadliwych komórek jest miękiszem, a liczba problemów rozwojowych jest zmniejszona..
Ta forma nazywa się mozaiką, ponieważ normalne komórki zmieszane z wadliwym. Nietypowe komórki z dodatkowym chromosomem są rozmieszczone losowo w całym ciele i nie ma możliwości ich usunięcia w jakikolwiek sposób.. Ta forma jest nieco łatwiejsza, czas życia jest dłuższy..
Podstawa patologii: czym jest wina genów?
Tylko jeden chromosom u dziecka ma wiele poważnych i poważnych konsekwencji dla zdrowia i życia. Wynika to z faktu, że we wszystkich komórkach istnieje uniwersalny system do odczytu informacji, a podczas podziału komórki - podwojenia informacji programowanych w cząsteczkach DNA. Jeśli występuje defekt co najmniej jednego genu - już wpływa on na organizm w postaci choroby lub problemu, oraz jeśli istnieje cały fragment dodatkowego chromosomu lub nawet całego chromosomu, występuje wiele wad rozwojowych i problemów zdrowotnych.
Chromosom 18 zawiera do 2,5% wszystkich informacji genetycznych o pracy organizmu, a zaburzenia reprodukowane z powodu tego zespołu są bardzo znaczące. Dodatkowe geny uszkodzonego chromosomu powodują syntezę nadmiaru białek, co prowadzi do nieprawidłowego rozwoju i funkcjonowania wielu narządów i układów dziecka. W zespole Edwardsa szkielet, niektóre strefy układu nerwowego, układu moczowo-płciowego i układu sercowo-naczyniowego są dotknięte, mają zarówno anomalie w strukturze, a następnie - jak płód rozwija się - w funkcjonowaniu.
W związku z tym główną i jedyną przyczyną tego zespołu jest tworzenie dodatkowego chromosomu w komórce seksualnej matki lub ojca. Przy wytwarzaniu komórek rozrodczych nie ma miejsca zwykły podział, a następnie podwojenie 23 par chromosomów do 46 sztuk (dla każdej identycznej kopii w parze), ale tylko 23 sztuki. Jeśli w procesie dzielenia, 18-ta para z jakiegoś powodu nie została prawidłowo podzielona, dwa chromosomy trafią natychmiast do komórki rozrodczej i nie będzie w niej 23 elementów, ale 24-d. Jeśli taka komórka doprowadzi do nowego życia (czy to plemnika ojca, czy jaja matki), doprowadzi to do pojawienia się dziecka z zespołem Edwardsa..
Czynniki ryzyka związane z zespołem Edwardsa
Aby komórki rozrodcze rodziców mogły dzielić się niepoprawnie, a ma się nadmiar zestawu chromosomów, potrzebne są różne negatywne wpływy, zarówno z zewnątrz, jak i wewnątrz ciała.. Prowadząc ich można rozważyć:
- wiek matki i ojca w momencie poczęcia. Istnieją rzeczywiste dowody na czynnik wieku na prawdopodobieństwo trisomii. Wraz z wiekiem zwiększa się liczba nieprawidłowych komórek w układzie rozrodczym, co jest związane z osłabieniem kontroli organizmu nad procesami podziału. Ryzyko posiadania dzieci z patologiami chromosomalnymi po 30 latach wzrasta 2-3 razy w porównaniu z młodym wiekiem, a po 40 latach szanse wzrastają o 6-8 razy więcej. Uzależnienie od wieku ojca nie jest tak oczywiste, ale defekty chromosomalne są również całkiem możliwe..
- mieć złe nawyki, szczególnie jeśli chodzi o uzależnienie od nikotyny i alkoholu. Powszechne zatrucia naruszają procesy podziału komórek, w tym narządów płciowych, co powoduje kilkakrotne zwiększenie ryzyka wystąpienia uszkodzonych plemników i jaj. Jeszcze bardziej niebezpieczne jest stosowanie środków odurzających i toksykomanii, które jeszcze bardziej naruszają procesy podziału komórki..
- brać trochę narkotyków, wpływające na procesy podziału komórki, nadmiar dawek i czas podawania, niepożądane połączenia leków podczas leczenia, powodujące ich toksyczne stężenie.
- infekcje somatyczne i narządów płciowych, które prowadzą do pokonania narządów rozrodczych i komórek, tworząc przewlekły przebieg, wpływają na procesy podziału (cytomegalia, opryszczka, brodawczak i inne).
- ryzyko zawodowe, różne rodzaje narażenia - jonizujące, magnetyczne i wiele innych. Wpływają one na proces podziału komórki, mają niszczący wpływ na DNA, prowadzą do powstawania wolnych rodników w komórkach i mutacji.
Ostateczne przyczyny i wszystkie czynniki ryzyka prowadzące do powstania komórek z trisomią można wyjaśnić jako odkrycia biologii molekularnej i genetyki. Wszystkie te czynniki tylko zwiększają ryzyko, ale nie mów, że takie dziecko się urodzi..
Nie podatny na tworzenie się trisomii w komórkach płciowych, czego dowodzi fakt, że w chwili narodzin w rodzinie jednego dziecka z zespołem Edwardsa szanse na urodzenie kolejnego dziecka wzrastają 200 razy w porównaniu ze średnią - z 0,04% do 2-3% lub więcej.
Charakterystyka zespołu Edwardsa w okresie noworodkowym
Chociaż dzisiaj możliwe jest zdiagnozowanie syndromu przed urodzeniem, patologię często stwierdza się już przy urodzeniu. Noworodek z podobną anomalią chromosomową ma typowe i wyraźne wady rozwojowe, ich obecność może pomóc w postawieniu diagnozy nawet bez dodatkowych badań chromosomów - analiza genetyczna (ale tak czy inaczej, bez tych danych diagnoza nie jest ostateczna).
Przy urodzeniu zidentyfikowano:
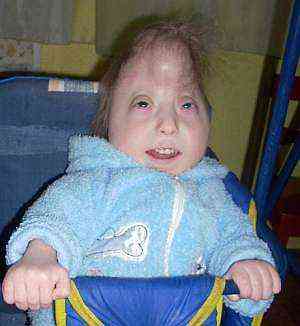 anomalie w strukturze kształtu czaszki. Szkielet z takim zespołem cierpi najsilniej - objawia się to przede wszystkim zmianą kształtu czaszki z utworzeniem dolichocefalu (ostro wydłużona, wąska czaszka - "wieża"). Taka anomalia nie jest typowa tylko dla tej patologii, ale wskazuje na istniejące nieprawidłowości rozwojowe. Najbardziej wyraźna zmiana w stosunkach według pomiarów w okolicy kości ciemieniowej i długości od mostka nosa do guzów potylicznych. Cięższą i cięższą anomalią czaszki może być małogłowie, nieproporcjonalnie mały rozmiar czaszki mózgu względem ciała..
anomalie w strukturze kształtu czaszki. Szkielet z takim zespołem cierpi najsilniej - objawia się to przede wszystkim zmianą kształtu czaszki z utworzeniem dolichocefalu (ostro wydłużona, wąska czaszka - "wieża"). Taka anomalia nie jest typowa tylko dla tej patologii, ale wskazuje na istniejące nieprawidłowości rozwojowe. Najbardziej wyraźna zmiana w stosunkach według pomiarów w okolicy kości ciemieniowej i długości od mostka nosa do guzów potylicznych. Cięższą i cięższą anomalią czaszki może być małogłowie, nieproporcjonalnie mały rozmiar czaszki mózgu względem ciała..- specjalnie zmodyfikowany kształt małżowiny usznej. Nieprawidłowa struktura uszu u noworodków jest silnie zaznaczona i jest obserwowana u 95% dzieci, zwłaszcza z pełnym typem zespołu. Przedsionki są znacznie niższe niż zwykle, stosunkowo ostro obniżone w stosunku do orbit, chrząstki są wypukłe i nie ma wyraźnie ukształtowanych loków. Płatek uszny i tragus są w powijakach lub są całkowicie nieobecne, przewód słuchowy ostro zwęża się i jedna czwarta dzieci może być całkowicie nieobecna..
- nienormalna struktura nieba za pomocą żuchwy (tworzenie się mikrognatiów). U noworodków, gdy podejrzewa się zespół Edwardsa, nie ma całkowitej akceleracji procesów podniebiennych w strefie górnej szczęki, które tworzą podłużną szczelinę. Mogą istnieć opcje z całkowitym rozłączeniem tylko miękkiego podniebienia lub częściowego zespolenia, czasami z przejściem do warg. Przy pełnej formie defektu, nierozłączność może być obustronna, górna warga jest rozszczepiona po obu stronach, powodując patologiczne szczeliny w szczęce i podniebieniu, co uniemożliwia dziecku całkowite zamknięcie ust. W przypadku poważnych patologii możliwa jest komunikacja między jamą ustną i jamą nosową, nawet jeśli usta są zamknięte. Częstotliwość występowania takich anomalii sięga 20-25% i więcej..
- Mogą być inne problemy. - gotyckie niebo (nadmiernie wysokie sklepienie kostne) i zmiany w strukturze żuchwy. Są zarejestrowane u około 70% dzieci. Typowy micrognathia - broda i żuchwa są mocno odsunięte, co uniemożliwia dzieciom całkowite zamknięcie ust i śliny wyciekającej z jamy ustnej. Przy takiej anomalii występują trudności z karmieniem, szczególnie poprzez stosowanie do piersi..
- deformacja stóp z formowaniem "kołysania się", charakterystyczne anomalie długości palców kończyn. Zmiana w stopie jest typowa dla 75% lub więcej dzieci na tle tego zespołu, następuje zmiana w położeniu kości - kostki, pięty lub łuski, co prowadzi do powstania silnie wystającej pięty pod nieobecność łuku stopy. Może być pochylony do przodu, dając stopom wyraz nóg z kołyskowego krzesła. Obserwuje się także zaburzenia równowagi długości nóg, szczególnie dużych. Jest krótszy niż drugi palec u nogi, jak widać po spłaszczeniu palców.
- dłonie w pozycji zginanej i zrosty między palcami, specjalne rysunki dermatoglificzne. Syndactyly (termin ten odnosi się do fuzji palców) jest wykrywany w 45% przypadków, zwykle jest palcami, ale może to wpływać na dłonie. W łagodnym stopniu skóra tworzy błony, a nadprzestrzenne kości mogą tworzyć się w ciężkich. Może być również zginać anomalie rąk - położenie palców w nienormalnej, stale napiętej pozycji, przykrywając małym palcem i kciukiem wszystkie pozostałe, mocno przyciśnięte do dłoni. Takie zaburzenia są typowe dla 90% dzieci z zespołem..
- Między innymi z trisomią typową specjalną wzory na palcach i dłoniach ze specjalną formą fałd skórnych. Wraz z rozwojem tego zespołu nieprawidłowości są typowe dla 60% dzieci. Są to bardzo częste łuki na klockach, brak fałdu skórnego pomiędzy skrajnymi i przedostatnimi paliczkami, rozwinięcie poprzecznego rowka - ma on termin "małpa".
- malformacje narządów płciowych. U chłopców jest to niedorozwój penisa i moszny, u dziewczynek wzrost wielkości łechtaczki. Anomalie są zwykle łączone z wadami układu moczowo-płciowego, ale najwyraźniej pozostałe znaki nie widzą.
 anomalie w strukturze kształtu czaszki. Szkielet z takim zespołem cierpi najsilniej - objawia się to przede wszystkim zmianą kształtu czaszki z utworzeniem dolichocefalu (ostro wydłużona, wąska czaszka - "wieża"). Taka anomalia nie jest typowa tylko dla tej patologii, ale wskazuje na istniejące nieprawidłowości rozwojowe. Najbardziej wyraźna zmiana w stosunkach według pomiarów w okolicy kości ciemieniowej i długości od mostka nosa do guzów potylicznych. Cięższą i cięższą anomalią czaszki może być małogłowie, nieproporcjonalnie mały rozmiar czaszki mózgu względem ciała..
anomalie w strukturze kształtu czaszki. Szkielet z takim zespołem cierpi najsilniej - objawia się to przede wszystkim zmianą kształtu czaszki z utworzeniem dolichocefalu (ostro wydłużona, wąska czaszka - "wieża"). Taka anomalia nie jest typowa tylko dla tej patologii, ale wskazuje na istniejące nieprawidłowości rozwojowe. Najbardziej wyraźna zmiana w stosunkach według pomiarów w okolicy kości ciemieniowej i długości od mostka nosa do guzów potylicznych. Cięższą i cięższą anomalią czaszki może być małogłowie, nieproporcjonalnie mały rozmiar czaszki mózgu względem ciała..Oprócz tych objawów istnieją również mniejsze oznaki i anomalie rozwoju dzieci, które mogą we wstępnej diagnozie tego zespołu. Ważne jest, aby pamiętać, że jeden z wymienionych objawów, a nawet dwa nadal nie mówią o diagnozie, istnieje wiele wad w zespole i mają różne kombinacje.
Jest ważnyWiększość opisanych objawów może występować jako pojedyncze warianty i nie potwierdza diagnozy, może być wyizolowanym wariantem nieprawidłowości podczas ciąży lub przejawem innych patologii. Diagnoza defektów zewnętrznych nie może być wykonana.!
Co więcej, na tle niekompletnych lub mozaikowych typów zespołu objawów zewnętrznych może być trochę jeden lub dwa, podczas gdy istnieją poważne defekty narządów wewnętrznych z częstymi zaburzeniami w chromosomach. To oni określają przyszłe prognozy dotyczące życia i przebiegu zespołu..
Rozwój dziecka: cechy zespołu Edwardsa
Jedynie pojawienie się wraz z pojawieniem się światła i przejawów wad rozwojowych, anomalia Edwardsa nie jest ograniczona, ponieważ rośnie i rozwija się, możliwe są przejawy nowych i cięższych odchyleń. Pierwszą z nich można zidentyfikować w ciągu kilku tygodni od chwili narodzin. Często po raz pierwszy można podejrzewać rozpoznanie mozaiki i niekompletnego typu patologii, jeśli jest kilka defektów zewnętrznych lub nie ma ich wcale.. Wyżej opisane defekty i zmiany zewnętrzne stają się ostrzejsze i bardziej wyraźne w miarę ich wzrostu, stopniowo pojawiają się nowe problemy i anomalie, które nie były zauważalne przy urodzeniu. Warto tutaj:
- Ostre i wyraźne opóźnienie w poziomie rozwoju fizycznego. Przy urodzeniu masa takich dzieci nie przekracza 2 - 2,5 kg, a powodem tego są defekty chromosomalne, które nie pozwalają na pełny rozwój systemów i narządów. Wraz z postępem rozwoju wzrost i masa ciała różnią się znacznie z miesiąca na miesiąc, a obwody piersi są również słabe. Rozmiary głów mogą sięgać normalnej lub wyższej, co może wskazywać na nagromadzenie nadmiaru płynu w jamie czaszki, tworząc wodogłowie.
- Problemy ze stopami. W miarę ich wzrostu powstaje stopa końsko-szpotawa z powodu anomalii w budowie kości i więzadeł, hipertonusa mięśni. Cierpienie i kontrola nad rozwojem stopy i napięcia układu nerwowego. Zagraża temu fakt, że dzieci zaczynają iść późno lub nie mogą tego zrobić w ogóle z powodu poważnego stanu ogólnego. Zewnętrznie, stopa końska objawia się poważną deformacją stopy i jej nienormalną instalacją w spoczynku.
- Zaburzenie budowy mięśni. Przy urodzeniu hipertoniczność jest typowa dla rąk, tworząc ich zginacze, ale gdy się rozwija, rozprzestrzenia się na sąsiednie grupy mięśniowe, prowadząc do artystycznych pozycji. Częściej jednak zmniejsza się napięcie mięśniowe, dzieci są powolne i atoniczne, kończyny ważą z biczami. Mogą również występować stany dystonii z ostrym skurczem poszczególnych grup mięśni na tle niedociśnienia innych. W takim przypadku ramiona mogą być zgięte, a nogi mogą się nadmiernie zgiąć. Prowadzi to do zaburzeń koordynacji i chaotycznych ruchów, dyslokacji i nieprawidłowej pozycji kończyn..
- Nietypowe reakcje układu nerwowego, problemy emocjonalne. Niektóre części mózgu u dzieci z zespołem Edwardsa są słabo rozwinięte, a zatem powstają problemy emocjonalne i hamujące, niewystarczające reakcje na środowisko. Zwykle wiąże się z ciałem modzelowatym i hipoplazjami móżdżku, które również tworzą upośledzenie umysłowe. Zewnętrznie objawia się to w nieobecnym spojrzeniu, braku emocji i niemożności nawiązania kontaktu, braku obiektów śledzących. Dzieci nie reagują na dźwięki z powodu problemów z uszami i układem nerwowym, takie anomalie są wykrywane w pierwszych miesiącach życia..
Generalnie, z powodu ciężkich i połączonych zmian, dzieci nie żyją do wieku większości.. Niektóre z nich w formie mozaiki mogą osiągnąć wiek nastolatków z głębokim stopniem głupoty i zewnętrznych przejawów wad rozwojowych..
Zespół Edwardsa Techniki diagnostyczne
Obecnie istnieją trzy typy zespołu diagnostycznego, które zasadniczo różnią się od siebie.. Biorąc pod uwagę, że patologia jest nieuleczalna i śmiertelna we wczesnym wieku, ważne jest, aby zwracać uwagę na diagnozę prenatalną, która nie pozwala na narodziny dzieci z tymi zaburzeniami. Pomocne będą również konsultacje genetyków w rodzinach, w których zdarzały się przypadki nieprawidłowości chromosomalnych.. Możesz wydać:
- Rozpoznanie komórek płciowych przed poczęciem. Niestety, ta metoda ma zastosowanie jest nadal bardzo ograniczona i tylko w protokołach IVF we wstępnym badaniu komórek płciowych. Jeśli jest to naturalna koncepcja, lekarze mogą przewidzieć większe ryzyko dla narodzin takich dzieci, ale to nic więcej niż przewidywania. Nie można na pewno wiedzieć, czy takie dziecko urodzi się, czy nie. Przeanalizowano historię rodzinną, biorąc pod uwagę wszystkie istniejące choroby, oszacowano czynniki ryzyka i przeprowadzono analizę genetyczną dla obojga rodziców, stosując zaawansowany sprzęt i testy. Poprzez określenie kariotypu rodziców można wyciągnąć pewne wnioski..
- Identyfikacja syndromu podczas ciąży. Wraz z rozwojem płodu istnieje kilka metod bezpośredniego lub pośredniego potwierdzenia zespołu Edwardsa: początkowo do tych celów stosuje się badanie ultrasonograficzne i badania krwi z badaniem konkretnych wskaźników, oraz przy wysokim ryzyku przeprowadzane są zabiegi inwazyjne - amniopunkcja lub biopsja kosmówki i biopsja kosmówki. Przeprowadzanie badań przesiewowych w ściśle określonych okresach, kiedy zmiana danych ultrasonograficznych i parametrów krwi będzie najistotniejsza.
Uzyskanie bezpośrednio komórek płodu lub jego krwi umożliwia prowadzenie badania jego chromosomów i potwierdzenie lub zaprzeczyć zespołowi Edwardsa. Pomaga to w podjęciu decyzji o przedłużeniu lub zakończeniu ciąży z przyczyn medycznych..
- Potwierdzenie syndromu po urodzeniu. Przeprowadza się go zgodnie z wynikami zewnętrznych danych w połączeniu z licznymi analizami genetycznymi potwierdzającymi obecność trisomii na 18. parze chromosomów. Ponadto ważne jest, aby ocenić stan dziecka i przewidzieć jego życie i stan zdrowia, aby zrozumieć, ile pomocy potrzebuje, aby przeżyć.. Bez opieki medycznej nad tym zespołem dzieci mogą szybko umrzeć w miejscu urodzenia w nadchodzących tygodniach.. Ultrasonografia i echokardiografia, radiografia i EEG, różne badania krwi i moczu, dodatkowe testy do MRI i CT.
Leczenie i rokowanie w zespole Edwardsa
Niestety, zespół ten jest nieuleczalny do tej pory, a lekarze mogą tylko pomóc dziecku lepiej radzić sobie z różnymi problemami poprzez interwencje chirurgiczne i leczenie zachowawcze.. Zwykle dzieci żyją do kilku miesięcy lub roku, tylko z częściową lub mozaikową formą mogą żyć dłużej. Śmierć wynika z wielu wad rozwojowych i nieprawidłowości w pracy narządów, a także z łączenia się z powikłaniami. Radykalnie poprawić defekty chromosomów w komórkach organizmu dziecka na obecnym etapie rozwoju medycyny jest niemożliwe.
Alyona Paretskaya, pediatra, recenzent medyczny