

Mukopolisacharydoza to cała gama genetycznie uwarunkowanych (warunkowanych) patologii, które pojawiają się w przypadku upośledzenia metabolizmu kwaśnych mukopolisacharydów (glikozoaminoglikanów).
Najczęściej z takim naruszeniem wpływa na szkielet i rozwój fizyczny jest opóźniony, a niektóre rodzaje patologii przejawiają zacofanie ze strony sfery intelektualnej. Ponadto występują uszkodzenia w układzie sercowo-naczyniowym, żołądkowo-jelitowym i nerwowym, w aparacie optycznym i skórze.
Leczenie mukopolisacharydozy jest objawowe, rokowanie jest ogólnie niekorzystne - upośledzenia postępują, ponieważ gromadzą się glukozaminoglikany. Pacjenci umierają wystarczająco wcześnie.
Dane ogólne
Mukopolisacharydozę badano stosunkowo niedawno - w XX wieku. Początkowo uważano ją za pojedynczą patologię, a następnie, dzięki różnorodności, zaczęli ją rozróżniać. Czynnikiem jednoczącym całą mukopolisacharydozę jest to, że kwaśne mukopolisacharydy gromadzą się w narządach i tkankach, a przyczyną tego naruszenia jest niższość niektórych enzymów komórkowych, które są włączone do genotypu i są dziedziczone.
Zwróć uwagęOrtopedzi leczą pacjentów z mukopolisacharydozą, ale wymagają także wysiłków klinicznych od kardiologów, okulistów, neurologów, otolaryngologów i kilku innych specjalistów..
Mukopolisacharydoza typu IH
Mukopolisacharydoza typu IH nazywana jest również zespołem Hurlera. Rozpoznano u 1 noworodka po 20-25 tys.
Pierwsze oznaki choroby pojawiają się w pierwszym roku życia dziecka, ale pełny obraz kliniczny może rozwinąć się dopiero o 2 lata..
Najbardziej typowymi objawami choroby są:
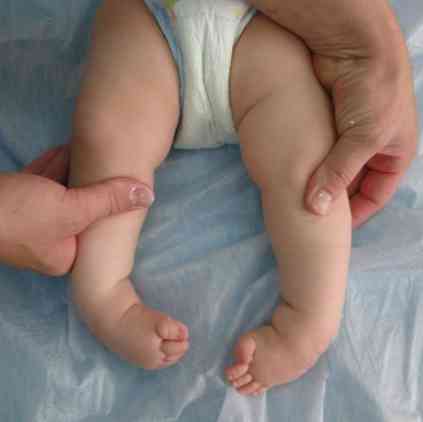 szorstkie kontury twarzy - bardzo typowe dla tej patologii;
szorstkie kontury twarzy - bardzo typowe dla tej patologii;- deformacja czaszki - staje się jak kil kilowy. Ten stan nazywa się scaracephalia;
- oddychanie odbywa się przez usta, ponieważ oddychanie nosa jest upośledzone z powodu zwiększenia się migdałków, jak również wewnątrzmaciczne zaburzenia rozwojowe w czaszce twarzy;
- deformacja kończyn i niektórych innych fragmentów szkieletu;
- opóźnienie wzrostu;
- skrzywienie kręgosłupa (objaw "grzbietu kota" w pozycji siedzącej);
- skrócenie szyi;
- nietypowy wysoki układ ostrzy;
- wybrzuszone do przodu niższe żebra;
- zwiększenie szerokości szczotek - podczas gdy przypominają one pazur z pazurem;
- rozwój przykurczów (zmniejszenie zakresu ruchu) przy stopniowym wsuwaniu różnych stawów w proces patologiczny. Po pierwsze, dotyczy to stawów łokciowych i barkowych, a następnie rozciąga się na stawy kolanowe, biodrowe i skokowe.
 szorstkie kontury twarzy - bardzo typowe dla tej patologii;
szorstkie kontury twarzy - bardzo typowe dla tej patologii;Zespół Hurlera charakteryzuje się specyficznym chodem - na palcach z wygiętymi nogami. Taka cecha wynika z ograniczenia ruchów spowodowanych przykurczami..
Badanie fizyczne ujawnia następujące:
- brzuch jest powiększony, jego ściana przednia u takich pacjentów jest osłabiona, dlatego często mogą rozwinąć się przepukliny pępkowe i pachwinowe;
- powiększona wątroba i śledziona;
- u mężczyzn często stwierdza się wodniak - jądro jest obrzęknięte;
- w niektórych przypadkach wykrywane są naruszenia koordynacji ruchów;
- ujawnił nadmierny wzrost puszystych włosów.
Gdy instrumentalne badanie jest określone przez:
- na elektrokardiogramie - oznaki rozlanego (rozległego) uszkodzenia mięśnia sercowego;
- upośledzenie wzroku i słuchu;
- oftalmoskopia ujawnia zmętnienie i powiększenie rogówki.
Najbardziej charakterystyczne są zmiany, które stwierdzają podczas badania rentgenowskiego:
- RTG kręgosłupa - pozwala zidentyfikować charakterystyczną deformację kręgosłupa. W tym przypadku kręgi mają kształt sześcianu, a ich krawędzie są zaokrąglone. Również oznaczona kifoza - skrzywienie kręgosłupa naprzód;
- RTG klatki piersiowej - zaznaczone pogrubienie fragmentów żeber przednich z równoczesnym ścieńczeniem ich odcinków tylnych, skracaniem i deformacją. Żebra w tym samym czasie nabierają szczególnego wyglądu i stają się podobne do łopaty lub szabli. Obserwuje się także zmniejszenie i pewne przesunięcie głów kości ramiennej;
- radiografia miednicy - objawia się zwężenie pierścienia miednicy, jamy panewki (te, w których są "podstawą" stawu biodrowego) są przekrzywione, a głowy kości udowych są zmniejszone;
- Zdjęcie rentgenowskie kości rurowych - widoczne jest przerzedzenie korowej (powierzchownej) warstwy kości i pewne rozszerzenie kanału szpikowego;
- Prześwietlenie kości ręki - pomaga wykryć niedorozwój dystalnych (paznokci) paliczków palców, skrócenie i poszerzenie środkowych i proksymalnych (tych bliżej dłoni) paliczków, a także kości śródręcza;
- RTG czaszki - wyraźnie słabo rozwinięte kości czaszki twarzy, czaszkowatość (zwężenie czaszki w niektórych miejscach) i makrocefalia (powiększenie czaszki).
U pacjentów z rozpoznaniem mukopolisacharydozy typu IH rozpoznano następujące zaburzenia, które można uznać za odrębne choroby:
- dystrofia pigmentu siatkówki - naruszenie jej rozwoju;
- jaskra - wzrost ciśnienia wewnątrzgałkowego;
- zatory w dnie oka;
- niedowład - zaburzenie ruchu;
- paraliż - całkowite zakłócenie aktywności ruchowej w pewnych obszarach układu mięśniowo-szkieletowego.
Z biegiem czasu pacjenci z diagnozą Hurlera rozwijają się i nieodwracalnie rozwijają upośledzenie umysłowe.
Mukopolisacharydoza typu I-S
Inną nazwą mukopolisacharydozy typu I-S jest choroba. Choroba została opisana dopiero w połowie XX wieku, chociaż pacjenci z charakterystycznymi przykurczami palców dłoni, zaburzeniami ruchowymi w dużych stawach i specyficznymi cechami twarzy spotykali się w praktyce klinicystów wcześniej. Objawy można łączyć z objawami zespołu Hurlera.
Ten rodzaj mukopolisacharydozy pojawia się w późniejszym wieku niż w przypadku mukopolisacharydozy typu IH, a jej przebieg jest korzystniejszy..
Charakterystyczną cechą mukopolisacharydozy typu I-S jest to, że do wieku 3-6 lat rozwój dzieci odpowiada normie wieku. Ponadto pojawiają się następujące znaki:
- przykurcze zgięciowe palców;
- ograniczenie rozciągania w dużych i średnich stawach - mianowicie łokciu, ramieniu i nadgarstku.
Naruszenie amplitudy ruchów części kończyn dolnych jest mniej wyraźne niż w innych typach opisywanej patologii.
Pełny obraz kliniczny tego typu mukopolisacharydozy ustalono na początku okresu dojrzewania.
W badaniu fizjologicznym (badaniu) stwierdza się:
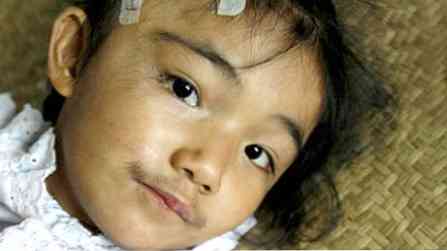 pacjenci nie są wysokie, ale mają silną budowę (krępy);
pacjenci nie są wysokie, ale mają silną budowę (krępy);- funkcje są raczej grube;
- muskulatura szkieletowa dobrze rozwinięta;
- obserwuje się nadmierne owłosienie (zwiększone owłosienie na ciele);
- skóra na palcach rąk (częściej) i stopy (rzadziej) jest pogrubiona i rozciągnięta;
- wielkość wątroby i śledziony z reguły nie uległa zmianie.
 pacjenci nie są wysokie, ale mają silną budowę (krępy);
pacjenci nie są wysokie, ale mają silną budowę (krępy);Ponadto ci pacjenci są uznani za naruszenia, które można uznać za odrębną chorobę:
- przepuklina pachwinowa lub pępkowa;
- zespół cieśni nadgarstka - rozwija się dzięki wyraźnej kompresji nerwu pośrodkowego. Objawia się przez atrofię (niedorozwój) mięśni grzbietowych (uniesienie w okolicy kciuka) i parestezje (zaburzenia czułości) w obszarze 3, 4 i 5 palców;
- zwężenie zastawki aortalnej - zwężenie aorty;
- niewydolność aorty;
- dystrofia pigmentu siatkówki;
- jaskra;
- zmętnienie rogówki.
Inteligencja pacjentów z rozpoznaniem mukopolisacharydozy typu I-S pozostaje prawidłowa, w przeciwieństwie do pacjentów z mukopolisacharydozą typu IH.
Z metod instrumentalnych najbardziej pouczająca jest radiografia. Identyfikuje zmiany, które są podobne do zmian w mukopolisacharydozie typu IH, ale są mniej wyraźne..
Mukopolisacharydoza typu II
Ten rodzaj mukopolisacharydozy nazywany jest również chorobą Huntera. Patologia najczęściej dotyka chłopców w wieku 2-3 lat.
Według objawów klinicznych choroba jest podobna do mukopolisacharydozy typu IH - podczas gdy występują:
- scariocephaly;
- chropowate cechy;
- niski ton głosu;
- trudności w oddychaniu - z powodu deformacji kości szkieletu twarzy.
Wraz z dalszym postępem choroby dołączają następujące objawy:
- zmniejszona inteligencja;
- koordynacja ruchów jest zepsuta;
- Nastrój u takiego pacjenta jest niestabilny - obserwuje się tak zwaną chwiejność emocjonalną - od ataków śmiechu do płaczu. Takie wahania nastroju mogą wystąpić w dowolnym momencie..
Wraz z postępem mukopolisacharydozy typu II pojawia się i zwiększa nieuzasadniona agresywność.
W przeciwieństwie do mukopolisacharydozy typu IH, złamania kręgosłupa w postaci kifozy nie są charakterystyczne, nie obserwuje się objawów "kotów".
Badanie fizyczne określa:
- wykryto niewielki wzrost śledziony i wątroby;
- na skórze grzbietu określa się określone guzki.
Kiedy badanie instrumentalne ujawniło:
- zmętnienie rogówki, które postępuje;
- rosnący ubytek słuchu.
Ze wszystkich instrumentalnych metod diagnostycznych najbardziej pouczająca jest radiografia różnych fragmentów układu mięśniowo-szkieletowego - czaszki, komórki rudnej, miednicy i tak dalej. Jednocześnie objawy radiologiczne są takie same jak w przypadku mukopolisacharydozy typu I-S.
Pacjenci z taką diagnozą są podatni na rozwój chorób układu oddechowego:
- zapalenie tchawicy;
- zapalenie oskrzeli;
- zapalenie płuc.
Należy zauważyć, że mukopolisacharydoza typu II może rozwinąć się w postaci dwóch opcji:
- niekorzystny (opcja A);
- korzystne (opcja B).
 Niepożądany wariant jest widoczny dzięki jasnemu obrazowi klinicznemu, przy jednoczesnym zauważeniu istotnych zaburzeń inteligencji. Fatalności mogą wystąpić w okresie dojrzewania.
Niepożądany wariant jest widoczny dzięki jasnemu obrazowi klinicznemu, przy jednoczesnym zauważeniu istotnych zaburzeń inteligencji. Fatalności mogą wystąpić w okresie dojrzewania.
Objawy o korzystnej odmianie nie wyrażone, powyższym naruszeniom może towarzyszyć lekkie opóźnienie umysłowe. Tacy pacjenci z reguły żyją do 30 lat, w niektórych przypadkach - i więcej.
Mukopolisacharydoza typu III
Mukopolisacharydozę typu III opisał amerykański pediatra Sanfillipo, więc patologia otrzymała inną nazwę - choroba Sanfillipo.
Choroba rozpoznawana jest u 1 dziecka na 100-200 tysięcy noworodków..
Dzieci z mukopolisacharydozą typu III w pierwszych latach życia rozwijają się zgodnie z normami wieku i można zaobserwować pewne objawy patologii - w szczególności trudności w połykaniu i niezręczny chód..
Począwszy od 3-5 lat dziecko zaczyna pozostawać w tyle za swoimi rówieśnikami w rozwoju fizycznym, a jednocześnie staje się apatyczne, obojętne na ludzi i zdarzenia.. Typowe objawy to:
- zaburzenia mowy;
- chropowatość cech;
- regularne nietrzymanie moczu (nie tylko nocne) i kał.
Gdy dziecko dorasta, upośledzenie umysłowe wzrasta i ostatecznie staje się głównym objawem tego typu mukopolisacharydozy..
Badanie fizykalne ujawniło:
- nieznaczne powiększenie wątroby i śledziony;
- jednolity wzrost dystrybucji włosów;
- przykurcz;
- opóźnienie wzrostu.
 Naruszenie narządu wzroku i układu sercowo-naczyniowego, które często występują w innych postaciach opisywanej patologii, dla tego typu mukopolisacharydozy nie są typowe.
Naruszenie narządu wzroku i układu sercowo-naczyniowego, które często występują w innych postaciach opisywanej patologii, dla tego typu mukopolisacharydozy nie są typowe.
Wyniki badania rentgenowskiego z opisaną patologią lub bez zmian patologicznych lub podobne do tych, które są wykrywane z mukopolisacharydozą typu I-S, ale mniej wyraźne.
Rokowanie dla tej choroby jest mniej korzystne niż dla innych typów mukopolisacharyozy - tacy pacjenci umierają w wieku 10 do 20 lat, powikłania infekcyjne stają się przyczyną śmierci..
Mukopolisacharydoza typu IV
Mukopolisacharydoza typu IV jest również nazywana chorobą Morkio (nazwa pochodzi od urugwajskiego pediatry, który ją opisał). Patologia występuje częściej niż inne typy mukopolisacharydozy - mianowicie u 1 dziecka na 40 tysięcy noworodków.
Dzieci z tą chorobą do 1-3 lat rozwijają się bez cech. Ale pojawiają się następujące naruszenia:
- dziecko znacząco pozostaje w tyle za swoimi rówieśnikami;
- w procesie rozwoju zauważył skrócenie szyi i tułowia;
- powstają różne kontrakty;
- skóra pogrubia;
- siła mięśni zmniejsza się;
- słuch staje się coraz gorszy;
- rysy twarzy stają się szorstkie;
- występują różne deformacje klatki piersiowej.
Także dla mukopolisacharydozy typu IV są naruszenia, które można uznać za odrębne patologie:
- koślawa deformacja kończyn - zakrzywienie w kierunku na zewnątrz;
- skolioza (skrzywienie kręgosłupa w płaszczyźnie bocznej) lub kifoza;
- przepuklina pachwinowa;
- przepuklina pępkowa;
- dystrofia rogówki.
Intelekt z tą formą mukopolisacharydozy uratowany.
Rozpoznanie opisanej patologii jest potwierdzone przez zdjęcie rentgenowskie, w tym przypadku najbardziej pouczające. Oto jego następujące typy:
- Zdjęcie rentgenowskie kręgosłupa - obrazy przedstawiają krzywiznę kręgosłupa w postaci kifozy i skoliozy, a także ekspansję i spłaszczenie kręgosłupa;
- radiografia miednicy - wykazano liczne deformacje pierścienia kości miednicy, nierówności jego konturów;
- RTG kończyn - wykrycie spłaszczenia głów kości udowej, wyraźne skrócenie kości przedramienia i deformacja kości stopy.
Pacjenci z mukopolisacharydozą typu IV żyją z reguły krócej niż 20 lat. Wyniki śmiertelne występują na tle wielu powiązanych chorób, które są ostatecznie skomplikowane z powodu niewydolności krążeniowo-oddechowej..
Mukopolisacharydoza typu VI
Inną nazwą mukopolisacharydozy typu VI jest choroba Maroto-Lamy (po nazwiskach dwóch francuskich lekarzy, którzy opisali tę patologię)..
Objawy kliniczne są w dużej mierze "odbijane" od objawów innych typów mukopolisacharydozy, ale najbardziej widoczne są:
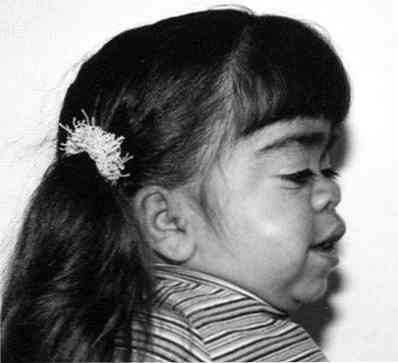 znaczące zgrubienie rysów twarzy;
znaczące zgrubienie rysów twarzy;- zauważalne opóźnienie wzrostu;
- skrócenie szyi - czasami wydaje się, że głowa pacjenta "siedzi" prawie na obręczy barkowej;
- deformacja klatki piersiowej.
 znaczące zgrubienie rysów twarzy;
znaczące zgrubienie rysów twarzy;Pacjenci tacy często mają przeziębienie - ten objaw pozwala podejrzewać tę konkretną, a nie inną postać mukopolisacharydozy..
Badanie fizyczne ujawnia następujące:
- wątroba i śledziona są często powiększane;
- przepuklina pachwinowa może być zdiagnozowana.
Intelektualnie dziecko rozwija się bez naruszeń - ma zdolność myślenia i rozwiązywania problemów intelektualnych..
Istnieją dwa warianty tego typu mukopolisacharydozy:
- klasyczny;
- mukopolisacharydoza z niewielkimi objawami klinicznymi.
W celu potwierdzenia diagnozy pomoże badanie rentgenowskie - obrazy są określane przez:
- deformacja kręgów - stają się prostopadłościenne lub klinowate;
- trójkątne odkształcenie pierścienia miednicy;
- niedorozwój (niedorozwój) i deformacja głów kości udowej;
- skrócenie kości strzałkowej.
Mukopolisacharydozy typu VII i VIII
 Mukopolisacharydoza tego typu jest mniej powszechna niż inne typy opisywanej patologii..
Mukopolisacharydoza tego typu jest mniej powszechna niż inne typy opisywanej patologii..
Mukopolisacharydoza typu VII występuje klinicznie jako choroba typu III, różnica między nimi występuje jedynie w wynikach badań biochemicznych.
Mukopolisacharydoza typu VIII jest objawowa i podobna do mukopolisacharydozy typu IV. Ale jest różnica w manifestacjach - jest to naruszenie rozwoju intelektualnego, którego nie obserwuje się w przypadku mukopolisacharydozy typu IV.
Diagnostyka
Przy diagnozowaniu mukopolisacharydozy opierają się na skargach, anamnestycznych danych, wynikach dodatkowych metod badania - fizycznych, instrumentalnych, laboratoryjnych. Można podejrzewać obecność mukopolisacharydozy przez charakterystyczne deformacje szkieletu, które "wpadają w oko", ale możliwe jest określenie rodzaju opisywanej choroby jedynie za pomocą dodatkowych metod badawczych. Główne metody to:
- radiografia;
- wykrycie glikozoaminoglikanów w moczu;
- oznaczanie aktywności enzymów w hodowlach komórkowych.
W przypadku klinicznych objawów zaburzeń narządów wewnętrznych stosuje się metody badania:
- elektrokardiografia;
- ultradźwięki wątroby i śledziony;
- elektroencefalografia;
- biochemiczny test krwi
i wiele innych.
Diagnostyka różnicowa
Rozpoznanie różnicowe (charakterystyczne) przeprowadza się pomiędzy różnymi typami mukopolisacharydozy.
Komplikacje
Najczęstsze powikłania mukopolisacharydozy to:
- przykurcz;
- niewydolność serca;
- niewydolność płuc.
Leczenie
Nie opracowano terapii mukopolisacharydozy, która może być stosowana w celu wyeliminowania choroby, a nie tylko jej objawów.. Tacy pacjenci otrzymują leczenie objawowe - może to być:
- konserwatywny - transfuzja krwi, terapia hormonalna, terapia witaminowa i tak dalej;
- operatywna - endoprotetyka główek kości udowej, naprawa przepuklin, interwencja chirurgiczna w celu wyeliminowania przykurczów i prawidłowych deformacji szkieletu i innych.
Ważne są:
- leczenie infekcji dróg oddechowych;
- korekcja wad wzroku i słuchu.
Zapobieganie
Ponieważ patologia powstaje z powodu zaburzeń genetycznych, nie ma konkretnych metod zapobiegania.. Jednak ryzyko zaburzeń genetycznych, które mogą prowadzić do rozwoju różnych typów mukopolisacharydozy, można zmniejszyć, jeśli kobieta otrzyma normalne warunki ciąży. To jest:
- odrzucenie złych nawyków;
- przynależność do pracy, odpoczynek, sen;
- optymistyczny nastrój.
Prognoza

Prognozy dotyczące mukopolisacharydozy są generalnie niekorzystne, dotyczy to wszystkich opisanych typów chorób. W procesie życia i rozwoju tkanek stopniowo gromadzą się produkty metaboliczne powstające w wyniku braku enzymów tkankowych. Prowadzi to do patologicznych zmian prawie wszystkich narządów i układów. Jeśli patologia się rozpocznie, śmierć może nastąpić nie tylko z powodu niewydolności sercowo-płucnej, ale także z powodu niewydolności wielosystemowej.
Terapia zachowawcza odgrywa rolę wspomagającą - stosowanie jakichkolwiek leków prowadzi do tymczasowej poprawy, ale nie jest w stanie zrekompensować braku enzymów tkankowych.
Kovtonyuk Oksana Vladimirovna, komentator medyczny, chirurg, lekarz konsultant